Desde que CRISPR/Cas9 hizo su aparición hace un poco más de una década se ha convertido en una herramienta de edición genética de uso cotidiano en investigación que facilita el trabajo en laboratorios alrededor del mundo. Sin embargo, antes de CRISPR/Cas9, ya existían tecnologías como las nucleasas de dedos de zinc (ZNFs) que fueron los pioneros en lograr la modificación del ADN. Asimismo, nuevas herramientas como Prime Editing y Base Editing son una demostración de que la historia de la edición genética es mucho más extensa de lo que normalmente se piensa y comprender las diferencias que presentan las herramientas que la componen permite la utilización de una variedad de estrategias para lograr diversidad de objetivos.
A medida que la edición genética se expandía en la industria y la salud, se hizo evidente una necesidad crítica: mayor precisión. Los científicos, impulsados por este desafío, desarrollaron una solución que superó los riesgos de la edición temprana, como las alteraciones no deseadas en el genoma. Esta solución consistió en el uso de endonucleasas, enzimas capaces de cortar el ADN en puntos precisos. Estas “superhéroes moleculares” permitieron una intervención más exacta y sentaron las bases de la edición genética moderna (Carroll, 2021).
Las primeras de estas herramientas fueron las nucleasas de dedo de zinc (ZNF), seguidas por los efectores similares a activadores de transcripción con nucleasas (TALEN) y más recientemente, el conocido sistema CRISPR/Cas9.
¿Cómo funciona ZFN?
Las ZFN (Zinc Finger Nucleases) son enzimas artificiales que combinan dos componentes: un dominio de unión al ADN (los “dedos de zinc”) y un dominio de corte (la nucleasa FokI). Los dedos de zinc están diseñados para reconocer una secuencia de ADN específica, mientras que la nucleasa es la que realiza el corte preciso en el genoma. Para funcionar, las ZFN trabajan en pares; cada una se une a una hebra de ADN adyacente. Cuando ambas se acoplan en su lugar, sus dominios FokI se unen para formar un dímero, lo que activa el corte y crea una rotura de doble hebra (DSB).
Una vez que el ADN se ha cortado, la célula tiene dos formas de reparar el daño. El mecanismo más común es la unión de extremos no homólogos (NHEJ), un proceso rápido pero propenso a errores. El segundo, y más preciso, es la reparación dirigida por homología (HDR), que utiliza una plantilla de ADN para asegurar una corrección sin fallos (Gupta & Musunuru, 2014)
¿Cómo funciona TALENs?
Los TALENs (Transcription Activator-Like Effector Nucleases) son otra herramienta de diseño que, al igual que las ZFN, utiliza un par de enzimas para cortar el ADN. Estas nucleasas están compuestas por dos partes: un dominio de unión al ADN derivado de bacterias del género Xanthomonas y un dominio cortador de la nucleasa FokI.Su mecanismo es muy similar al de las ZFN. Cada uno de los dos TALENs reconoce una secuencia específica en el ADN. Cuando se unen a sus respectivos sitios, sus dominios FokI se juntan, formando un dímero que crea una rotura de doble hebra (DSB) en el ADN. Al provocar esta rotura, los TALENs permiten a los científicos introducir mutaciones, ya sea eliminando o insertando segmentos de ADN en puntos específicos del genoma (Gupta & Musunuru, 2014).
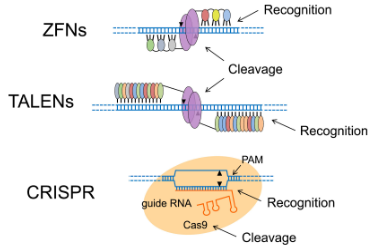
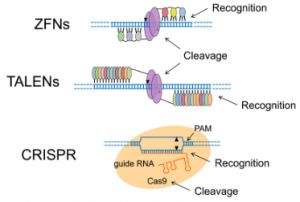 Figura 1. Mecanismos de acción de ZFNs, TALENs y CRISPR. La figura ilustra las tres herramientas de edición genética. Las ZFNs y TALENs usan dominios de unión al ADN para guiar la nucleasa FokI y cortar ambas hebras. Por su parte, CRISPR usa la proteína Cas9 y un ARN guía para reconocer la secuencia objetivo y cortarla, siempre que haya una secuencia PAM adyacente. Créditos: Adaptado de Carroll (2021).
Figura 1. Mecanismos de acción de ZFNs, TALENs y CRISPR. La figura ilustra las tres herramientas de edición genética. Las ZFNs y TALENs usan dominios de unión al ADN para guiar la nucleasa FokI y cortar ambas hebras. Por su parte, CRISPR usa la proteína Cas9 y un ARN guía para reconocer la secuencia objetivo y cortarla, siempre que haya una secuencia PAM adyacente. Créditos: Adaptado de Carroll (2021).
En comparación con herramientas más recientes como CRISPR/Cas9, tanto las ZFN como los TALENs presentan fortalezas importantes, así como desafíos que limitan su uso generalizado. Por un lado, los TALENs destacan por su alta especificidad y menor toxicidad, lo que les confiere precisión en la edición genética y reduce el riesgo de efectos no deseados en regiones no objetivo. Además, son más flexibles y más fáciles de diseñar que las ZFN, gracias al código modular de reconocimiento por nucleótido único, lo que permite ensamblarlas rápidamente con alta eficiencia (Gupta & Musunuru, 2014). Sin embargo, ambos sistemas requieren ingeniería proteica específica para cada blanco genético, lo cual implica mayor tiempo, esfuerzo y costos, lo que demuestra una barrera significativa frente a CRISPR/Cas9, que simplemente requiere cambiar la guía de ARN (sgRNA) para redirigir la nucleasa (Li et al., 2020).
CRISPR ha demostrado así ser una opción más práctica, eficiente y escalable para la mayoría de aplicaciones de edición génica, aunque los TALENs y ZFN aún conservan su lugar en escenarios donde la precisión y el control son críticos.
No obstante, la evolución de la edición genética no se detuvo en CRISPR. De esta tecnología se derivaron herramientas más sofisticadas, como Base Editing y Prime Editing, diseñadas para superar algunas de las limitaciones del sistema convencional.
Base Editing es capaz de modificar una sola de las “letras” del ADN (las bases nitrogenadas A, C, G y T) sin necesidad de cortar ambas hebras. Este enfoque evita el daño en la molécula y permite una corrección segura de mutaciones puntuales. La técnica, que funciona con editores de citosina (CBE) y de adenina (ABE), puede, por ejemplo, cambiar directamente una C por una T, o una A por una G (Komor et.al., 2016).
Por otro lado, Prime Editing, llamado por muchos como “procesador de texto del ADN”, puede realizar ediciones más complejas. Esta herramienta no solo localiza el sitio exacto a editar, sino que también lleva consigo una “guía especial” que contiene la información correcta. Esto permite insertar, eliminar o reemplazar grandes fragmentos de información genética con una precisión notable, como si se cortara, pegara y reemplazara texto en un documento (Anzalone et.al., 2019).
¿Y existen variantes de la herramienta CRISPR?
CRISPR a diferencia de lo que muchas personas piensan es una familia de herramientas diversas, cada una con distintas funciones. Por ejemplo, se puede mencionar a Cas12a (conocida antes como Cpf1), la cual se diferencia de Cas9 al poder trabajar con regiones del ADN a las que Cas9 no alcanza porque Cas12a utiliza secuencias TTTV (en las cuales V puede ser A, C, o G). Asimismo, Cas12a realiza cortes cohesivos, lo que quiere decir que forma pequeñas colas de nucleótidos sin aparear en cada extremo lo que permite que estas se unan más fácilmente. Cabe destacar que, Cas12a tiene la capacidad de cortar múltiples sitios haciendo uso de varios ARN guías en una sola reacción y su menor tamaño es favorable cuando se requiere el uso de vectores virales (Zetsche et al., 2015). Si bien, puede parecer que Cas12a presenta más ventajas que Cas9, lo cierto es que esta última sigue siendo la nucleasa CRISPR con más estudios y variantes que permiten el mejoramiento de su especificidad y eficiencia (Hsu et al., 2014)
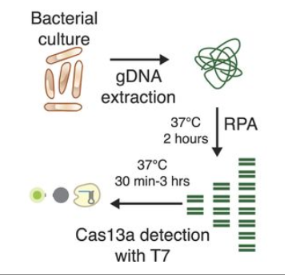 Figura 2. “Esquema de cómo se usa SHERLOCK para distinguir las cepas bacterianas con un conjunto de cebadores universales del gen 16S rRNA V3 RPA.” Créditos: Adaptado de Gootenberg et al., (2017).
Figura 2. “Esquema de cómo se usa SHERLOCK para distinguir las cepas bacterianas con un conjunto de cebadores universales del gen 16S rRNA V3 RPA.” Créditos: Adaptado de Gootenberg et al., (2017).
Por otra parte, se tiene a Cas13a la cual no corta ADN sino ARN. Esta tecnología es guiada por un ARN CRISPR para el reconocimiento de secuencias de ARN mensajero o viral y una vez se une a la secuencia se quiere cortar, Cas13a activa su dominio ribonucleasa HEPN (Higher Eukaryotes and Prokaryotes Nucleotide-binding Domain), el cual es un dominio proteico cuyo uso es cortar ácidos nucleicos. En el caso de Cas13a, HEPN degrada al ARN objetivo pero también corta a otros fragmentos de ARN cercanos aunque estos no coincidan con la secuencia que se buscaba. Aunque esto podría verse como un problema, se hizo uso de esta propiedad para diseñar sistemas de detección sensibles como SHERLOCK, el cual consiste en la mezcla de Cas13a, un ARN guía y un ARN reportero que produce un cambio de color o fluorescencia cuando se lo corta. Esta plataforma de diagnóstico molecular permite la detección de material genético con alta especificidad y precisión de microorganismos como virus o bacterias (Abudayyeh et al., 2016; Gootenberg et al., 2017).
Similarmente a Cas12a, Cas14 es una nucleasa CRISPR bastante compacta por lo que se la utiliza para aplicaciones que involucren su entrega a través de vectores virales de tamaño reducido. Lo que la hace diferente a Cas14 es que puede atacar a una cadena sencilla de ADN y comparte la característica de Cas13a de cortar moléculas cercanas una vez que se ha unido a la secuencia objetivo lo que abre la posibilidad de desarrollar plataformas de diagnóstico molecular de alta sensibilidad y especificidad para microorganismos patógenos (Harrington et al., 2018).
Del mismo modo, se tiene a CasΦ que fue descubierta en bacteriófagos de genomas muy extensos y es de dimensiones más compactas respecto a Cas9 y Cas12a, aproximadamente 70 kDa (la mitad de Cas9) lo que facilita su introducción en vectores como virus adenoasociados. CasΦ funciona con un ARN guía simple y corta ADN de doble cadena, lo que se traduce en que tiene la capacidad de edición genética pero con menor carga genética para ser entregada (Pausch et al., 2020). Por otra parte, se puede observar que Cas12a y CasΦ cuentan con funciones similares al poder ser empaquetadas en vectores virales por tener un tamaño más reducido que Cas9. No obstante, CasΦ al ser más reciente cuenta con menos estudios sobre su eficiencia y tampoco se han publicado estudios que respalden la posibilidad de evasión a sistemas inmunológicas bacterianos.
¿A qué conclusiones podemos llegar?
Es importante recalcar que herramientas como TALENs y ZFNs ofrecen alta especificidad y han sido validadas clínicamente manteniéndose vigentes en ciertas aplicaciones donde se requiere máxima precisión y se conoce ampliamente su perfil de seguridad. Además, las herramientas de Base Editing y Prime Editing demuestran que la evolución de CRISPR es imparable y cada vez se encuentran soluciones más especializadas para cada reto. Del mismo modo, el sistema CRISPR se basa en una diversidad de nucleasas, cada una con sus ventajas y desventajas, algunas con orígenes en bacteriófagos como CasΦ, así como variantes que atacan el ARN y no el ADN, en el caso de Cas13a, lo que permite a los investigadores escoger la estrategia más adecuada para optimizar la eficiencia y minimizar riesgos. Por otra parte, el fomentar el conocimiento sobre las diferentes aplicaciones y funciones que tienen cada de estas herramientas es de interés para la sociedad en general debido a la posibilidad no sólo de desarrollar terapias génicas de manera más segura para enfermedades hereditarias, sino para sectores como la agricultura en la que se puedan desarrollar cultivos más resistentes a condiciones climáticas adversas y con más aporte nutricional sin depender de una sola tecnología.}
Bibliografía
- Abudayyeh, O. O., Gootenberg, J. S., Konermann, S., Joung, J., Slaymaker, I. M., Cox, D. B. T., … Zhang, F. (2016). C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science, 353(6299), aaf5573. https://doi.org/10.1126/science.aaf5573.
- Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., Chen, P. J., Wilson, C., Newby, G. A., Raguram, A., & Liu, D. R. (2019, October 21). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature, 576(7785), 149–157. https://doi.org/10.1038/s41586-019-1711-4
- Carroll, D. (2021). A short, idiosyncratic history of genome editing. Gene and Genome Editing, 1, 100002. https://doi.org/10.1016/j.ggedit.2021.100002
- Fridovich-Keil, J. (s. f.). Gene editing. Encyclopaedia Britannica. https://www.britannica.com/science/gene-editing
- Gootenberg, J. S., Abudayyeh, O. O., Lee, J. W., Essletzbichler, P., Dy, A. J., Joung, J., … Zhang, F. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science, 356(6336), 438–442. https://doi.org/10.1126/science.aam9321
- Gupta, R. & Musunuru, K. (2014). Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. Journal of Clinical Investigation, 124(10), 4154–4161. https://doi.org/10.1172/JCI72992
- Harrington, L. B., Burstein, D., Chen, J. S., Paez-Espino, D., Ma, E., Witte, I. P., … Doudna, J. A. (2018). Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science, 362(6416), 839–842. https://doi.org/10.1126/science.aav429
- Hsu, P. D., Lander, E. S., & Zhang, F. (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell, 157(6), 1262–1278. https://doi.org/10.1016/j.cell.2014.05.010
- Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., & Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature, 533(7603), 420–424. https://doi.org/10.1038/nature17946
- Li, H., Yang, Y., Hong, W., Huang, M., Wu, M., & Zhao, X. (2020). Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduction and Targeted Therapy, 5, Article 1. https://doi.org/10.1038/s41392-019-0089-y
- Pausch, P., et al. (2020). CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science, 369(6501), 333–337. https://doi.org/10.1126/science.abb1400
- Urnov, F., Rebar, E., Holmes, M., Zhang, H., & Gregory, P. (2010). Genome editing with engineered zinc finger nucleases. Nature Reviews Genetics, 11(9), 636–646. https://doi.org/10.1038/nrg2842
- Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell, 163(3), 759–771. https://doi.org/10.1016/j.cell.2015.09.038